病態
遺伝性痙性対麻痺 (hereditary spastic paraplegia: HSP) は、緩徐に進行する下肢の痙縮 (筋肉のつっぱり) と筋力低下により歩行障害をきたす様々な病気の総称です。家族性痙性対麻痺 (familial spastic paraplegia: FSP) と呼ばれることもありますが、最近では、遺伝性であることを明らかに示すためにHSPと呼ばれることがほとんどです。HSPでは脊髄の錐体路、後索、脊髄小脳路と言われる部位の神経細胞が系統的に障害されます [1, 2]。
遺伝形式からは、常染色体優性遺伝性 (AD-HSP)、常染色体劣性遺伝性 (AR-HSP)、X連鎖性 (XL-HSP)、ミトコンドリア遺伝性 (mt-HSP) に分けられます。その頻度は、AD-HSPが多く、AR-HSPは少なく、XL-HSPとmt-HSPは稀です。
原因遺伝子そのものや原因遺伝子座 (遺伝子の場所) が判明した順にHSPの病型に対してSPGナンバーがつけられており(SPG1〜SPG91)、これまでのところ70を越える原因遺伝子が判明しています(https://neuromuscular.wustl.edu/spinal/fsp.html)。HSPは同じSPGナンバーの遺伝子変異であっても異なる症状を示すことがあり、逆に異なる遺伝子変異であっても同じ症状を示すことがあるので、遺伝子診断により病型を確定することが必要です。さらに、遺伝性がはっきりしない場合でもHSP原因遺伝子変異を認める症例があること、SPGナンバーがついていない病気でも痙性対麻痺の症状を認めることに注意が必要です。
患者数
欧米のHSPは、10万人あたり4.3〜9.8人程度であると報告されています [3]。我が国では、特定疾患臨床調査個人票の解析から10,487人の脊髄小脳変性症患者の4.7%を占めるとされています [4]。現在では30,000人を越える脊髄小脳変性症患者が特定疾患として登録されていますので、HSP患者数は1,500名程度と推測されます。
2006年に厚生労働科学研究費補助金難治性疾患克服研究事業 運動失調に関する調査研究班により ”多施設共同研究体制 (Japan Spastic Paraplegia Research Consortium (JASPAC)” が立ち上げられ、これまでHSPの全国的な臨床調査と遺伝子解析が行われてきました [5]。
図1に我が国のHSP病型頻度を示します。AD-HSP 211家系中SPG4が最も多く43%、次いでSPG3A 5%、SPG31 5%、SPG10 2%でした。AR-HSP 174例では、SPG11 6%、PLAN、ARSACS、CHS、SPG15、SPG46がそれぞれ2%でした [6]。AR-HSPはAD-HSPに比べて多様性に富むことがわかっています。AD-HSP、AR-HSP共に未だ原因遺伝子がわかっていない症例が多いので、今後、新しいHSP原因遺伝子の発見によりSPGナンバーは増えていくと思われます。
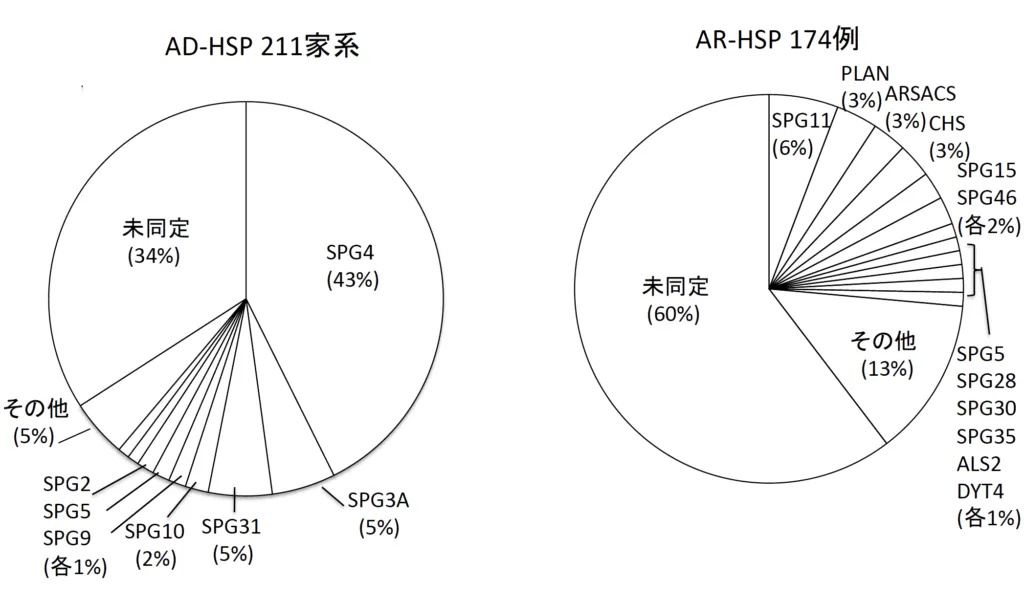
図1. 我が国のHSP病型頻度
原因
タンパクの機能解析から推測される分子機序からは、脂質/ステロール代謝、ミエリン/オリゴデンドロサイト、ミトコンドリア、ゴルジ-小胞体輸送、小胞体-ゴルジ膜輸送、軸索輸送、微小管、小胞体、ライソゾーム/エンドソーム オートファゴソームの多様な障害が考えられています [7]。
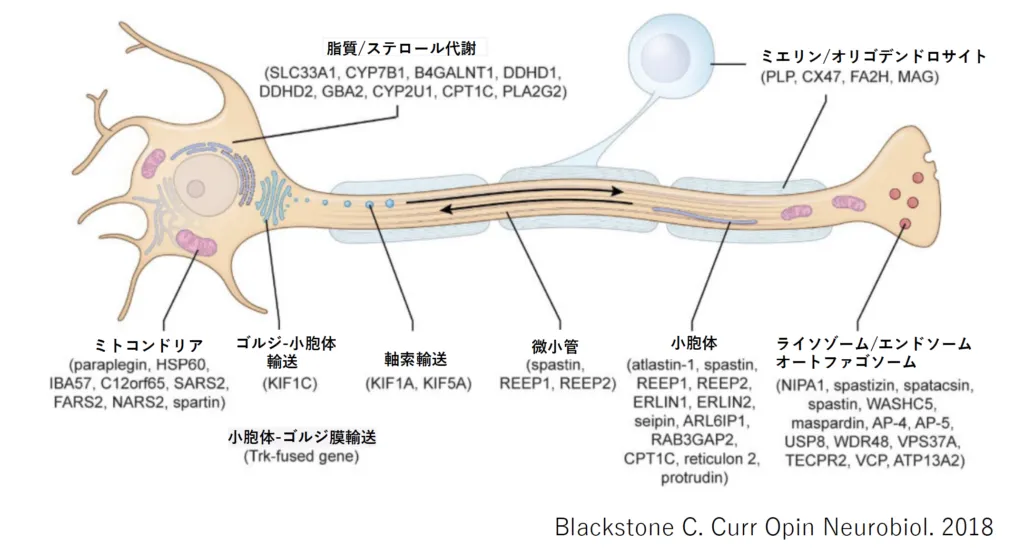
図2. HSPにおける障害部位とその原因遺伝子
症状
発症年齢は、乳児期、小児期、成人期、老年期とさまざまです。多くの患者さんの初発症状は歩きにくさを感じることです。処女歩行が遅れたり、運動会でのかけっこが遅かったりすることもあります。また、靴のつま先がすり減りやすいことに気がついている方もいます。
臨床的には、随伴症状の有無により純粋型と複合型に分けられます。純粋型は通常、下肢の痙縮、筋力低下、腱反射亢進、Babinski徴候 (病的な反射) といった痙性対麻痺のみの症状を呈します。複合型はこれらの症状に加えて、末梢神経障害 (足のしびれ感)、小脳失調 (ふらつき、呂律が回りにくい)、精神発達遅延 (知的障害)、認知症、けいれん、難聴、網膜色素変性症 (夜盲)、錐体外路症状 (体がこわばる、動作が遅い)、魚鱗癬 (皮膚が厚い) などの症状を伴います [1, 2]。
症状は緩やかに進行し、欧米の報告では、発症から22年で半数の患者は歩行補助具が必要になり、37年で約1/4の患者が車椅子を必要とします。高齢発症であるほど経過は重症で、独歩不能になるまでの経過が早いとされています。病気の重症度には病気になってからの期間が最も強い影響を及ぼしていることがわかっています。
治療
現在のところ下肢痙縮についての対症療法のみであり、抗痙縮薬の内服、ボツリヌス毒素治療、バクロフェン髄注療法が行われています。排尿障害については、対症的に内服薬治療や間欠的自己導尿などを考慮します。
継続的なリハビリテーションを行うことはきわめて重要です。特に2023年10月1日に厚生労働省から医療用HALの保険適用の通知が出されましたので、HSPに対してのHAL治療の効果が期待されるところです。
私たちは、モデル動物を用いてHSP治療法開発の研究を継続しており [8]、できるだけ早い時期に臨床研究に繋げたいと考えています。
解説:瀧山 嘉久
山梨大学名誉教授/笛吹中央病院院長
引用文献
- [1] McDermott CJ, Shaw PJ: Hereditary spastic paraplegia. In: Handbook of Clinical Neurology. Vol 82. Motor neuron disorders and related diseases (ed by Eisen AA, Shaw PJ), p198-211, Elsevier Science, Amsterdam, 2007.
- [2] 瀧山嘉久:遺伝性痙性対麻痺. Annual review神経 (柳澤信夫ほか編), 198-211, 中外医学社, 2008.
- [3] Finsterer J, Loscher W, Quasthoff S, et al: Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci 318: 1-18, 2012.
- [4] Tsuji S, Onodera O, Goto J, et al. Sporadic ataxias in Japan: a population-based epidemiological study. Cerebellum 7: 189-197, 2008.
- [5] 瀧山嘉久:本邦の痙性対麻痺に関する全国多施設共同研究体制 (JASPAC). 脊椎脊髄 27: 737-745, 2014.
- [6] 瀧山嘉久:遺伝性痙性対麻痺 (HSP) の病態とわが国における研究の現状. 全国脊髄小脳変性症 (SCD)・多系統萎縮症 (MSA) 友の会ニュース 234: 15-20, 2018.
- [7] Blackstone C. Converging cellular themes for the hereditary spastic paraplegias. Curr Opin Neurobiol 51: 139-146, 2018.
- [8] 瀧山嘉久:遺伝性痙性対麻痺 (HSP) の最新情報:本邦の分子疫学と治療法開発に向かって. 全国脊髄小脳変性症 (SCD)・多系統萎縮症 (MSA) 友の会ニュース 234: 15-20, 2018